Production Technology
Progress made in removing water soluble organics from GOM produced water
Removing water soluble organics from produced water can now be achieved with minimal corrosion and no scaling.
Paul R. Hart, Baker Petrolite Corp., Houston
Oilfield produced water contains a diverse mixture of compounds that varies from formation to formation. Of particular importance are the organic compounds classified as “Oil and Grease” (O&G) by the Clean Water Act.1
O&G must be removed to meet environmental, political and operational goals. Excessive O&G in re-injected water can foul the equipment or the formation. Discharged water must meet legal or contractual standards. The O&G in water discharged to the Gulf of Mexico (GOM) is limited to a 29-mg/L yearly average.
THEORY AND DEFINITIONS
For the purpose of discharge, O&G is a legal category. It comprises the residue of compounds that extract into n-hexane from water at pH < 2, after boiling away the solvent.2 The EPA uses ALOLA (a lot of long acronyms) to categorize different portions of this material.
The term “Water Soluble Organics” (WSO) is often used to describe the polar, non-strictly-hydrocarbon portion of the O&G compounds, which adsorb onto silica gel.3 While this could, in general, be any polar material, in the GOM’s produced waters, they are comprised largely of medium-chain carboxylate salts. The levels are especially high in newer deepwater production. A complete analytical flow diagram is shown in Fig. 1.
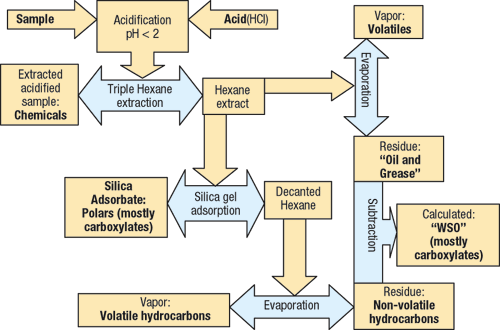 |
Fig. 1. Analysis flow diagram.
|
|
Process control analyses offshore are conducted using procedures simpler than the EPA’s. Typically, a sample is extracted only once rather than three times. This makes a big difference on compounds that partition to both phases. Extracting 50% of something three times in series leaves only 1/8th of it behind. The extract is then typically quantified photometrically, usually by IR, rather than gravimetrically by evaporation.
Because the polar O&G compounds’ chemical nature in GOM produced water has been so consistently carboxylate based, a simpler alternative to removing the polars with silica has been to omit the acidification, so the carboxylate salts do not extract. However the tests are done, the field results are calibrated to official EPA results, and appropriate correction factors, assuming constant composiions are used to evaluate performance.
The amount of WSO in O&G has no legal significance. It is measured as a diagnostic tool. It is the portion not removable by treatments used to remove particulate dispersions. If it exceeds the O&G limit, it is impossible to comply by using conventional clarification treatments, alone.
A wide spectrum of carboxylic acids can be found in petroleum naphtha. They have the general formula CnH2(n-z)O2, or equivalently, HCnH2(n-z)COOH, where the oxidation of hydrocarbon to acid has removed one hydrogen pair (or added one “double bond equivalent,” DBE), forming a “naphthene,” thus the term, “naphthenic acids.” Within a certain range of carbon numbers (n), the anions or conjugate bases of these acids partition as ionic salts into the produced water at the system pH, but they partition as acids into the O&G extraction solvent at the more acidic test pH.
Other types of polar organics can interfere with this simple interpretation in different ways, depending on the analytical protocol employed. If a non-evaporative technique, such as solution IR or UV, is used to measure the extract, small, non-ionic polar compounds will also be included as WSO. This includes compounds, such as butanol or acetaldehyde, that partition to both oil and water, regardless of the pH.
Conversely, some ostensibly non-WSO compounds, not polar enough to adsorb onto silica, such as benzene and small alkyl ethers, also partition to both oil and water, and so are truly water-soluble organics. Most such compounds, however, are also volatile enough that the concentrations present will evaporate with the hexane in an EPA protocol analysis.
While most hexane extractable silica adsorbers are (true) water soluble organics, most (true) water-soluble organics are not included in the O&G at all. Species, such as methanol and polyols, amines and polyamines (used as water clarifiers, corrosion and hydrate inhibitors), humic-type acids, and small carboxylic acids like acetic, do not extract into hexane at pH 2. There are more inclusive measures of dissolved organics, such as Total Organic Carbon (TOC). TOC is measured by quantifying the CO2 generated by total oxidation. This is often 10 times higher than the O&G content of produced water. TOC, and the closely related Chemical and Biological Oxygen Demands (COD/BOD), are of concern in the treatment of wastewater but have not been of concern in the disposal or reuse of produced water, because they do not (generally) cause fouling, and they do not count as O&G. They do, however, contribute to oxygen depletion or hypoxia, and regulating their discharge in the GOM’s hypoxia zone was once considered.4
Moreover, some materials that go into hexane at pH 2, but stay in water at the higher system pH, are not only not carboxylates, they are not even truly water-soluble. Despite the WSO nickname, there is nothing about their definition that requires they be truly in solution, if by that is meant organic molecules individually surrounded by water molecules. They could be stably emulsified hydrocarbons, part of a group of organic molecules not individually surrounded by water but surrounded as a group by water.
Small enough groups of molecules, though technically emulsified, behave in some ways like solutions. These are often given the special status of colloidal solutions or microemulsions. Molecular aggregates smaller than 1 µm diameter are generally considered colloidal, although they might contain as many as a billion molecules. For example, 100 million cetane molecules would create a 0.2 µm (200 nm) diameter particle, smaller than the wavelength of visible light and therefore non-reflective – invisible even at high concentration.
Polar organics concentrate on the surface of dispersed particles, often stabilizing the emulsion. Geometrically, the surface area of a given volume increases proportionally as the particle size drops. So, smaller particles tend to contain a larger percentage of polar material. Ultimately, a (dry) micelle is a group of molecules, all of which are on the surface and are polar organics, but none of which are true WSOs.
Carboxylate properties. The concentration of an organic in produced water does not depend on its solubility in water per se, but on its relative solubility in water and oil, or partition function. The partition coefficient is typically expressed as Log Ph/w, the logarithm of the ratio of its concentration in aliphatic hydrocarbon (e.g. heptane) to its concentration in water. Thermodynamically speaking, Log P is proportional to the free energy of phase transfer. The contribution of each molecular fragment to Log P is thus additive. For carboxylates, the difference between the contribution of the anion, – COO –, and the acid, -COOH, to LogPh/w is about 17. That is, for a given molecule, the anion partitions to water 1017 times more than the acid.5
The actual partition coefficient for a given carboxylic acid, however, even as the acid, is a function of both its concentration and the pH. In the oil, the acids form dimers, their heads held together tenaciously with complementary hydrogen bonds. These dimers are about 50 times more lipophilic (Log Ph/w 1.7 higher) than the free-headed monomers. Dimer formation depletes the monomer in the oil and pulls more of the acid remaining in the water into the oil. Because it is a bimolecular reaction, the concentration of dimer is proportional to the square of the concentration of monomer in the oil. Thus, the greater the concentration of the acid in the water, the greater the proportion of it partitions to the oil.6
Dimerization is an effect peculiar to carboxylic acids, regardless of size or surfactancy. It is not related to the higher aggregations of the longer chain, more surface-active acids into micelles in the oil or water, above a certain critical concentration. In a crude oil-water production system, these surfactant acids mostly reside at the oil-water droplet interfaces, rather than as free anions or oil phase dimers. Submicron (colloidal) oil droplets stabilized with such surfactants can appear (visually) to be dissolved in the water, but in reality they constitute a dispersed organic fraction that is far easier to remove than ruly dissolved anions. This is discussed in more detail in the next section. The partition values for anion, acid, and dimer for a series of carboxylates is shown in Table 1.
| TABLE 1. Log P(Heptane/ Water) for H(CH2)nCOOX |
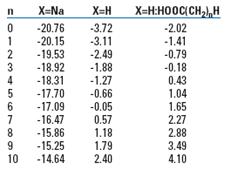 |
|
Below about pH 9, there is little countervailing complexation in the water. But above that pH, the anions predominate and cling to the few residual acids in multi-molecular complexes that shift the partition of the acid increasingly to the water, with increased concentration.7 Few GOM produced waters, however, are above pH 9.
Acid dimers in the oil are also less interfacially active than the free-headed monomers. The rate at which the partition re-equilibrates with a change in the water’s pH is limited by the rate at which the reactive head group in the oil contacts the water. Thus, once in the oil, the dimerized carboxylic acids are much slower to return to the interface than they were to leave it when water conditions change.8
Emulsion properties. Whereas truly dissolved organics like carboxylates must be desolubilized stoichiometrically to be removed, dispersed organics can be flocculated en masse.This can be a far more efficient intervention, as it involves interacting only with the surfaces of the particles.
For example, on a 100-µm droplet, only 1 out of every 3,000 or so molecules of cetane would be on the surface. Thus, 3,000 ppm of such oil particles could be removed with a mere 1 ppm of the right surface treatment. That is the magic of water clarification chemicals, how so little can do so much. But 1-µm droplets of the same 3,000-ppm oil would require 100 ppm to treat the whole surface, and 0.1-µm droplets would require 1,000 ppm for the same surface coating. The magic is lost as size decreases, and disappears completely as true (molecular) solution approaches.
The actual amount of treatment required depends not only on the surface area, but also on the magnitude of the stabilizing forces arrayed there and the efficiency with which the treatment counteracts them. One way that the polar organics at the surface impart stability is by creating a net charge repulsion that inhibits particle contact. In particular, the longer chain, surface-active, carboxylic acids noted earlier concentrate at this interface and dissociate to anions, depending on pH, leaving behind a mutually repulsive, anionic (negative) charge. Generally, nitrogenous bases from the asphaltene fraction also contribute a partially neutralizing, cationic (positive) charge, also dependent on pH.
The amount of net charge repulsion is thus a function of pH. As the pH is lowered (greater acidity), the carboxylic acids become less dissociated (more protonated) and so impart less charge. At the pH of the “isoelectric point,” charge repulsion disappears, and the particles coagulate (though they may not coalesce).
While such protic coagulants are universal, they also react with water (to form hydronium) and with metal surfaces. This renders them inefficient and corrosive. More efficient, but less universal, coagulants include binding polyvalent metal salts, polymeric ammonium salts, and micellar ammonium surfactants. The specificity of these traditional cationic coagulants renders their efficacy dependent on the anionicity of the particle surface. Thus a change in pH, which changes the surface charge, will require a change in the choice of coagulant to maintain the same removal of dispersed oil.
PROCESS DESCRIPTION
The removal of emulsified oil from water is typically a multi-step process. First, the mixed oil and water production is separated into two bulk phases, generally with the aid of chemical emulsion breakers, both “reverse breakers,” for oil-in-water, and “obverse breakers” for water-in-oil. Then the residual emulsion in the effluent water is further destabilized chemically, and the oil that breaks out is separated gravitationally or centrifugally. The residual emulsion from this separation might be treated further chemically before the oil droplets are reduced in density by attaching them to gas bubbles. These bubbles float the oil to the surface to be skimmed. Finally, the water might be passed through filters or absorbers of various media prior to discharge.
Virtually all the hydrocarbons are conveniently recovered in this manner. However, such traditional oil-water separation methods cannot remove truly water-soluble compounds that still count as O&G. In the GOM, the best coagulant and flocculant type clarifiers have been able to remove essentially all the dispersed organics, but only the 10% to 20% of the WSO associated with microemulsions.
Chemical solutions. A variety of chemical treatments has been proposed for removing various instances of apparently water-soluble organics. None of these has proven entirely satisfactory. Many are specific to certain types of dissolved or colloidal organics, and they do not apply to aqueous naphthenic carboxylate ions that ply the GOM’s produced water.
For example, both colloidal solutions of hydrocarbons and micellar solutions of soaps occur in certain industrial applications. These can be destabilized, and the components separated, with a combination of polyvalent metal hydrates and water soluble cationic polymers.9,10 Similarly, some emulsions or microemulsions are irremovable with conventional cationic clarifiers, because they are stabilized by cationic, amine-based corrosion inhibitors or overdoses of recycling, cationic clarifiers. These can be destabilized, and the components separated, with a combination of water-soluble anionic polymers and nonionic coalescing surfactants.10 Truly soluble aromatic hydrocarbons, such as benzene, partitioned into water from benzene-rich condensates, can be salted out of water and into oil using certain non-alkaline hydroxide salts of acidic metals.12 Truly-dissolved, reactive, polar, aprotic organics, such as aldehydes and ketones, can be kept from extracting into the analyte solvent by reacting them with hydrophilic reagents like diamines, alkanolamines, hydroxylamine, hydrazine and sodium bisulfite.13, 14
Historically, the only cost-effective means of moving truly dissolved carboxylate ions into oil has been by converting them to the oil-partitioning acid form by protonating them using a stronger mineral acid.15, 16 These acids are typically ~10N solutions, added in the 200-to-800-ppm range. The concentration needed will depend on the amount of buffering by the conjugate anions in the produced water, both those removable and those not (like acetate). The strong treatment acid must be added to the free water in mixed production, ahead of the primary separator, where there is bulk oil for the weak WSO acid to go into for removal.
However, secondary problems caused by these mineral acids often preclude their use. These include the health, safety and environmental hazards of handling mineral acid, increased corrosion of storage and processing equipment, scaling of processing equipment and production lines, and reduced effectiveness of conventional water clarifiers at removing the dispersed O&G. For example, both the hydrohalic acids (HF, HCl, etc.) and the hydrocarboxy acids [H2(CO2)x, i.e. formic and oxalic] are volatile, toxic and corrosive enough to harm human health and downstream distillation processes; monoprotic oxy acids (HNOx, HClOx) have dangerous oxidation potentials; and nonvolatile, multi-protic oxy acids (HxSOx, HxPOx) form insoluble scale deposits with common cations. In particular, sulfuric acid forms barium sulfate (BaSO4), phosphoric acid forms calcium phosphate [Ca10(OH)2(PO4)6], and phosphorous acid forms calcium phosphite (CaPHO3).
The scaling acids have been the least risky and most economical to use. Various attempts have been made to overcome the scaling, including the conjunctive use of scale inhibitors or the incorporation of weaker descaling acids into the formulation.17 These attempts have generally failed to meet expectations. The reason is that the same hydrophobic carboxylates being removed from the water readily adsorb onto the surface of the precipitated scale particles, using the same binding sites that the hydrophilic, carboxylate-based, anti-scaling compounds would have used. This renders the anti-scalent substantially ineffective, and the oil-coated, untreatable scale goes on to foul critical control valves and deposit in the produced oil line, restricting its flow.
One effective way to minimize scale is to use phosphorous acid (H2PHO3) at low pH.17 The pKa of the second, weaker proton is 6.6. As the pH is pushed below that, the conjugate is increasingly the bi-phosphite (HPHO3 – ), which does not precipitate. This, however, requires several times more acid, since only half the protons are active and many more are needed to further lower the pH. Both the phosphite anion and the lower pH are more aggressively corrosive to the carbon steel production lines.
Still, there is a window of use. The solubility of calcium phosphite (CaPHO3) at 120°F is about 100 ppm.18 If, for example, a pH of 5.6 could be achieved with 600 ppm of H2PHO3, there would only be enough PHO3= for 90 ppm of CaPHO3, and thus no scale. But to pH 6.6, only 120 ppm could be fed until scaling ensued.
Generally, because of the high buffering of carboxylate-infested waters, it takes more phosphorous acid to achieve a given pH than the amount that can be fed without scaling, corroding, or going broke. It typically takes about twice that much.
Another problem with acids is incompatibility with existing phase separation treatments. When fed ahead of the separator, they change the nature of the emulsions entering it. The lower the pH is driven, the greater the cationizing effect on the surface of the oil-in-water emulsion particles. This reduces the charge differential between the particles and the clarifier, which is a critical driving force in the clarification process.
Moreover, the finely precipitated, oil-wet, scale particles stabilize emulsions and create parasitic surfaces that consume clarifier. Complex, expensive adjustments are often needed to keep the dispersed O&G from going up as the dissolved O&G is driven down.
A less corrosive, non-scaling approach is to complex the carboxylate with a lipophilic hydro-ammonium salt, such as a medium-chain, trialkyl ammonium hydrochloride (R3NHCl).19 These can form a hydrophobic, hydrogen-bonded complex with a medium-chain carboxylate anion:
| |
RCOO – + R3NHCl " RCOO:HNR3 + Cl – |
(Eq. 1) |
But this complex is still less hydrophobic than the simple acid dimmer, and the stoichiometry is no better than the same reaction without the extra mass and expense of the amine in the middle:
| |
2RCOO – + 2HCl " RCOOH:HOOCR + 2Cl – |
(Eq. 2) |
Scaling, corrosion and emulsification are avoided, but in return for less removal, and only by adding five times the active treatment at twice the cost per active treatment.19
BEST TECHNOLOGY
The latest method developed for removing carboxylate-anion-type WSO from produced water does not use strong, corrosive acids, does not create scale, and is compatible with conventional water clarifier treatments.20
Chemistry. This method employs certain “activated hydrophilic acids” (AHAs), alone or in combination with anionic polymers, to reduce the total O&G. AHAs are a type of weak organic acid. They are non-toxic and readily biodegradable.
The hydrophilic group keeps them in the water, even at pH < 2, so that they are not counted as O&G themselves. It also renders them non-volatile, and so less dangerous and corrosive to downstream distillation equipment.
The activating group makes them just strong enough to efficiently protonate the naphthenate anions in the water. Although several hundred times less acidic than their closest competitor, phosphorous acid (H2PHO3), AHAs have proven just as effective at equal dosages.
Although adding the AHA forms no emulsion-stabilizing scale, the lower pH can adversely affect the existing clarifier treatment. This can partly offset WSO reduction in the O&G. This occurs because the AHA neutralizes the charge on the native anionic surfactants (the same type of carboxylate it is removing) without affecting the native cationic surfactants (basic nitrogen compounds). This reduces or even flips the prior charge on the emulsion from negative to positive. Standard cationic reverse breakers used to remove these emulsions are then no longer as complementary, and may even go from being destabilizing to restabilizing.
Adding an anionic polymer, along with the AHA, instead of or in addition to the standard cationic reverse breaker, overcomes this problem and minimizes total O&G.20 The anionic polymer can be added to the water at any point prior to the final clarification unit. It can be added separately, which allows more flexibility, or as part of a single product formulation, which is simpler.
Corrosion. It is not just the acidity of an acid that makes it corrosive; it is the solvating power of its conjugate anion.21 One key advantage of the AHA desolubilizers, compared to their mineral acid counterparts, is that for the same amount of acidity, they are far less corrosive in the two relevant environments – neat acid on the 316 stainless steel used to transport, store and feed it, and dilutions in water as high as 1,000 ppm on the 1018 carbon steel used in the production piping, into which it would be fed. Table 2 compares the least corrosive mineral acid in use, phosphorous, to the leading AHA, in both inhibited and uninhibited formulations, for both these environments. The AHA is appreciably less corrosive.
| TABLE 2. Corrosivity of desolubilizing acids |
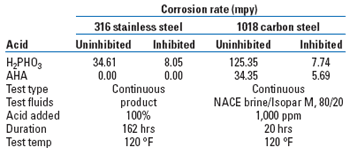 |
|
Consider also that the testing at 1,000 ppm on 1018 carbon steel coupons was done in synthetic brine. This exaggerates the actual corrosion, since, where they would really be added, the pH reduction is buffered by the carboxylates being treated, as well as the larger pool of parasitic, irremovable carboxylates. The standard synthetic brines do not include this pool of organic anions, and so the pH is far more depressed.
The actual amount of pH depression needed to effect treatment is typically only one pH unit, say, from 8 to 7 or from 7 to 6. In some cases, a drop of a few tenths of a pH unit is sufficient. Depression of pH to an excessively corrosive level is avoided. In any event, any acid would be fed to the same target pH. The difference in corrosivity at that pH is due to the more aggressive way that the phosphite anion dissolves iron compared to the AHA.
Scale potential. The most dramatic difference between phosphorous and AHA is that the Ca complex of a typical AHA remains soluble up to 15,000 ppm at 82°F, 150 times more soluble than the 100-ppm limit of phosphorous acid, and well in excess of any amount that would ever need to be fed. Other metal salts are even more soluble, as shown in Table 3.
| TABLE 3. Aqueous solubility of AHA metal salts |
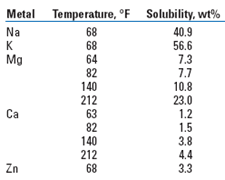 |
|
WSO REMOVAL
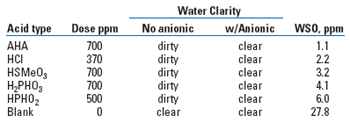
The ability of various materials to remove real WSO from real produced water was tested by simulating the entire production process, including all chemical treatments, in a series of glass bottles. A large number of experimental treatments, of several different chemical types, reflecting various theoretical mechanisms of actions, was tested. All, and only those acids capable of protonating naphthenic carboxylates, were effective, and effective roughly in proportion to their ability to do so. Some examples are listed in Table 4. Note that all the acids had the same directional effect on the emulsified oil.
| TABLE 4. Chemical response |
 |
|
Fig. 2 compares phosphorous acid to AHA proton for proton, first counting both of the mineral acid’s ionizable protons, and then counting just the stronger one, compared to the single ionizable proton of the AHA. At first, the greater ionization potential of the stronger proton on phosphorous acid (PA 1-H) gives it a slight, ~10% advantage. But the other proton (PA 2-H) is mostly useless and eventually parasitic, as additional strong protons are consumed reprotonating the weaker sites. The AHA’s molar efficiency is ultimately superior.
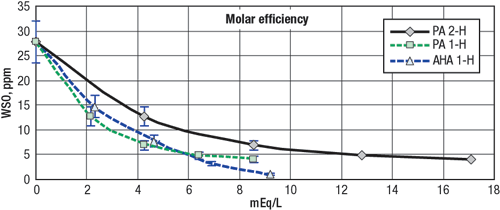 |
Fig. 2. WSO removal per acid proton.
|
|
Field use. AHA desolubilizers were added to actual produced water on various GOM platforms. The treatment scheme is shown in Fig. 3. Platform A had used 300-ppm phosphorous acid to reduce the produced water’s pH from 7 to 6.4. This reduced the WSO from 45 ppm to 20 ppm, but deposits of calcium phosphite (CaPHO3) chronically plugged the oil production line. A trial of an AHA desolubilizer produced a similar reduction in pH and of WSO with no trace of scale, Table 5.
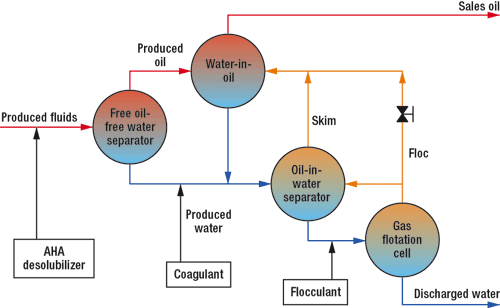 |
Fig. 3. Simplified process flow and treatment schematic.
|
|
| TABLE 5. Platform A: Trial 1 |
 |
|
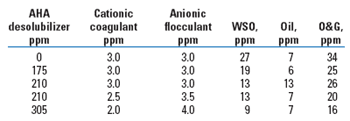
In this trial, the AHA reduced the WSO from 47 to 22. The insoluble oil increased from 9 to 11. The net effect was a 41% reduction, comparable to that seen with the phosphorous acid. The AHA was brought back after a series of emergency shutdowns caused by phosphite scaling of control valves. A switch to the AHA desolubilizer stopped the scaling completely, and even cleared up the residue. O&G results are shown in Table 6.
| TABLE 6. Platform A: Trial 2 |
 |
|
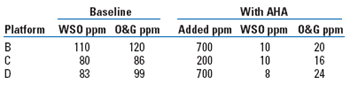
In this trial, after reducing O&G from 34 to 25 ppm, further reductions in WSO were met with a corresponding increase in insoluble oil, from 6 to 13 ppm. Feeding a few ppm of additional anionic flocculant at the expense of the cationic coagulant rebalanced the charge and brought the insoluble oil back down to 7 ppm. AHA desolubilizers were first made available commercially in April 2002. Results of the first three commercial applications are summarized in Table 7.
| TABLE 7. Commercial applications |
 |
|
The AHA feed rates listed are based on water production, and are higher where the water production is lower. Feed rates based on oil are more consistently in the 50-to-150-ppm range. The amount of oil matters more than water, because the carboxylates being treated in the water are actually coming from the oil.
In the application on Platform B, no free water was even present to treat. So the demulsifier injection point was moved ahead of the desolubilizer injection point, to create a free water phase for injection. This worked.
In all these applications, greater efficiency was obtained by treating only the wells with the highest concentrations of carboxylates, not the highest amounts, as is often done to minimize feed points. This created local concentrations as high as 1,300 ppm. It is thought that the higher concentrations enhanced the partitioning for a given pH reduction, and after the commingling water was diluted to a higher pH, the oil was separated before it could re-equilibrate.
CONCLUSIONS
Methods now exist for removing carboxylate anion-type WSO from GOM produced water without the corrosion and scale problems caused by mineral acids. Activated hydrophilic acids (AHAs), added at about 200 to 800 ppm (based on water) to mixed oil and water production, alone or together with anionic polymers at about 1 – 5 ppm, have been universally effective at transferring these organics from the water into the oil. 
LITERATURE CITED
1 US Federal Register 40 CFR 401.16
2 US Environmental Protection Agency, “Method 413.1 and Method 413.2”, methods for chemical analysis of water and wastes, 3rd edition, Environmental Monitoring Systems Laboratory-Cincinnati (EMSL-Ci), EPA-600/4-79-020, 1983.
3 US EPA, “Method 1664 revision A: n-hexane extractable material and silica gel-treated n-Hexane extractable material by extraction and gravimetry,” National Service Center for Environmental Publications, EPA-821-R-98-002, 1999.
4 Hill, T., acting branch chief, Region 6, U.S. EPA, “Discharge of produced water to the hypoxia zone,” Offshore Operators Committee General Meeting, New Orleans, Dec. 1, 2004.
5 Rekker, R. F. and R. Mannhold, Calculation of Drug Lipophilicity, VCH Verlagsgesellschaft mbH, 1992.
6 Goodman, D. S., “The distribution of fatty acids between n-Heptane and aqueous phosphate buffer,” JACS, 80, 3887; 1958.
7 Smith, R. and C. Tanford, “Hydrophobicity of long-Chain n-Alkyl carboxylic acids, as measured by their distribution between heptane and aqueous solutions,” Proc. Nat. Acad. Sci., USA, 70-2, 289; February 1973.
8 Hart, P. R., “Methods for inhibiting corrosion,” US Patent 6,103,100, Aug. 15, 2000.
9 Hart, P. R., “Method of removing benzene from petroleum desalter brine,” US Patent 5,236,591, Aug. 17, 1993.
10 Brown, W. and M. Trevino, “Wastewater organic acid removal process,” US Patent 5,395,536, March 7, 1995.
11 Bourg, D., “Demulsification of oil and water emulsions,” US Patent 6,153,656, Nov. 28, 2000.
12 Hart, P. R., “Method for Removing Soluble Benzene from Effluent Water”, US Patent 5,282,974 (Feb 1, 1994)
13 Morrow, L., M. Nellie, M. Kirkpatrick and A. Hossein, “Methods of removing water soluble organics from oil process water”, US Patent 5,804,078, Sept. 8, 1998.
14 Rush, T., “Low molecular weight amines and amine quaternaries for the removal of soluble organics in oil field produced water,” US Patent 4,354,477, Oct. 11, 1994.
15 Bellos, T., R. Greenlee and F. Welge, “Method of removing water-soluble organics from process water,” US Patent 4,818,410, April 4, 1989.
16 Ruebush, A., S. Davis, Jr., and V. Norviel, “Process for removing water soluble organics from produced water,” US Patent 4,839,054, June 13, 1989.
17 Bellos, T. and G. Noelken, “Method of removing water soluble organics from oil process water with an organic acid and a mineral acid having a plurality of pKa’s,” US Patent 5,853,592, Dec. 29, 1998.
18 Matlach, B., “Solubility of calcium phosphite”, Baker Petrolite Analytical Services Report 0102A198, April 14, 2001.
19 Means, C. and M. Squicciarini, “Organic ammonium salts for the removal of water soluble organics in produced water”, US Patent 6,159,379; Dec. 12, 2000.
20 Hart, P. R., “Removal of water solubilized organics,” US Patent 6,695,968; Feb. 24, 2004.
21 Reed, C. A., M. Juhasz, S. Hoffmann, K. C. Kim and E. Stoyanov, “The strongest isolable acid,” Angewandte Chemie, International Edition, Vol. 43, Issue 40 , pp. 5352-5355; Oct. 11, 2004.
|
THE AUTHOR
|
 |
Paul R. Hart is a development associate with Baker Petrolite’s Production Optimization Technology group in Sugar Land, Texas, where he leads product development for water clarification and oil sands applications. Prior to that, he led the Phase Separation research group for the Hydrocarbon Process division of BetzDearborn (now GE). Mr. Hart holds 44 published patents (eight currently pending). He has authored or co-authored 46 technical papers. He attended the University of Washington in Seattle, 1973 – 1977, receiving a BS degree in chemistry with honors.
|
|
|

